暂态测试方法.pdf
- 文件大小: 569.44KB
- 文件类型: pdf
- 上传日期: 2025-08-25
- 下载次数: 0
概要信息:
1
四、电化学暂态测量方法
2
暂态过程
暂态是相对于稳态而言的,当极化条件改变时,电极会从一个稳态向另
一个稳态转变,要经历一个不稳定的、变化的过渡阶段,这个阶段称为
暂态。在暂态阶段,电极电位、电极界面状态、扩散层的浓度分布都可
能发生变化,所以暂态系统要比稳态复杂的多。
暂态过程类似于电工学中所讨论的过渡过程,对于一个包括有电容和电
感的电路,当电路由一种稳定状态改变至另一种稳定状态时,一般来说
是不能瞬间完成的,需要一个转变过程,电极从开始极化到进入稳态同
样需要经过一定的时间才能达到,把这一过程称为暂态过程。
在暂态过程中,组成电极过程的各基本过程如溶液中离子的电迁移过程
、双电层充放电过程、电化学反应过程、传质过程等均处于暂态,描述
电极过程的物理量如电极电位、电流密度、双电层电容、浓度分布等都
可能随时间发生变化,导致暂态过程十分复杂。
3
暂态特点
1. 暂态过程具有暂态电流,即双电层充电电流;
极化电流包括两个部分:
一部分电流用于电化学反应,符合Faraday定律,即每电化当量的电化学反
应产生的电量为一个Faraday,称为法拉第电流(Faradaic current) if,或者
电化学反应电流;
另一部分电流用于双电层充电,称为双电层充电电流(double-layer charging
current) ic,或者称为电容电流(capacitive current)。
电极等效电路图
R1
Rf
Cd
i
iC
if
f ci i i 稳态: ic=0
暂态:ic变化 4
暂态特点
[ ( )
( )d z d
c d z
d C E E dCdq dE
i C E E
dt dt dt dt
双电层充电电流ic为
式中,取负号是因为规定阴极电流为正,Cd为双电层电容,E为电极电位,
Ez为零电荷电势。
式中第一项为电极电位改变时引起的双电层充电电流,第二项为双电层电容
改变时引起的双电层充电电流。
当电极表面发生吸脱附时,双电层电容Cd将发生剧烈的变化,由第二项引起
的充电电流可以达到很大的数值,常常形成吸(脱)附电流峰。因此,利用非法
拉第电流可以研究电极表面活性物质的吸脱附行为,还可以测定电极的双电
层电容和真实表面积。
5
极化前,电极处于平衡状态,双电层荷电状态保持稳定。
当通电后,由于电化学反应速度慢于电子运动的速度,被恒定电流驱使到达电
极界面的三对正电荷和电子中,只有一对相结合发生还原反应,另外两对排布
在电极界面两侧,改变了双电层的荷电状态,增大了电极的极化。这时,总电
流中的2/3为双电层充电电流,电极处于暂态。
S M
平衡态
M+
M+
M+
e-
e-
e- (M++e-→M)
S M
暂态
(M++e-→M)
(M++e-→M)
S M
M+
M+
M+
e-
e-
e-
暂态
S M
M+
M+
M+
e-
e-
e-
(M++e-→M)
(M++e-→M)
(M++e-→M)
稳态
电极通电前后电极/溶液界面处的变化情况
暂态特点
6
S M
平衡态
M+
M+
M+
e-
e-
e- (M++e-→M)
S M
暂态
(M++e-→M)
(M++e-→M)
S M
M+
M+
M+
e-
e-
e-
暂态
S M
M+
M+
M+
e-
e-
e-
(M++e-→M)
(M++e-→M)
(M++e-→M)
稳态
电极通电前后电极/溶液界面处的变化情况
随着电极极化进一步增大,电化学反应可在更高的速度下进行,被恒定电流
驱使达到电极界面的三对正电荷和电子中,可有两对相结合发生还原反应,
另外一对排布在电极界面两侧,进一步改变了双电层的荷电状态,增大了电
极的极化,这时,总电流中的1/3为双电层充电电流,电极仍处于暂态。
更大的电极极化可使三对正电荷和电子相互结合发生还原反应,全部电流都
用于电化学反应,双电层充电电流下降为零,电极达到稳态。
暂态特点
2
7
2. 在暂态下,电极附近液层中的反应离子浓度、扩散层厚度及浓度梯度等均随
时间变化,反应粒子浓度不仅是空间位置的函数,而且是时间的函数。
( , )C f x t
C
x
常数
暂态特点
平板
电极
表面
液层
中反
应物
浓度
分布
8
在同一时刻,浓度随离开电极表面的距离而变化,在离电极表面同一距离处
,浓度又随时间的变化而变化;随时间的推移,扩散层的厚度越来越大,扩
散层向溶液内部发展,当到达对流区时,建立起稳态扩散,扩散层厚度达到
最大,扩散层内离子浓度不再随时间而变化。可见,非稳态扩散过程比稳态
扩散过程多了时间这个影响因素。因此,可以通过控制极化时间来控制浓差
极化。通过缩短极化时间,减小或消除浓差极化,突出电化学极化。
暂态特点
9
1.暂态法适合研究快速电极过程,测定快速电极反应的动力学参数,能
够大大提高测量上限。
暂态方法的优点
暂态法的测量上限,交换电流密度i0<1A/cm2,电极反应速度常数k≤10-2cm/s
;稳态法的测量上限约为i0≤10-3A/cm2,k≤10-5cm/s
采用暂态法可以通过缩短极化时间,使扩散层有效厚度变薄,从而大大减少
浓差极化的影响,使电子转移步骤成为控制步骤,因此有利于测定快速电极
反应的动力学参数。
2.对于快速电极反应,浓差极化对电极过程影响较大,难以用稳态法测
量。采用暂态方法,可以控制条件,使得电极表面附近反应离子浓度
基本不变,忽略浓差极化的影响。
a. 采用小幅度测量信号,使电极极化很小(η<5~10mV)
b. 缩短单向极化时间。
10
3.暂态法可以研究反应产物在电极表面累积的情况,或者是电极表面在
反应时不断受到破坏的电极过程,稳态方法不行。
暂态方法的优点
暂态法测量时间短,液相中的杂质粒子往往来不及扩散到电极表面,因此,
可以研究界面结构和吸附现象,也可以研究电极反应的中间产物及复杂的电
极过程。
暂态法还可以研究那些表面状态变化较大的体系,如金属电沉积、金属腐蚀
过程等。这些过程反应产物能在电极表面上积累或者电极表面不断受到破坏
,稳态方法很难得到重现性比较好的结果。
11
暂态过程的分析方法
由于暂态系统是随时间而变化的,相当复杂,因此常常将电极过程用等效
电路来描述。
每个电极基本过程对应一个等效电路元件。只要得到等效电路中某个元件
的数值,也就知道了这个元件所对应电极基本过程的动力学参数。这样,
就将对电极过程的研究转化为对等效电路的研究。也就是说,将抽象的电
化学反应,用熟悉的电子电路来模拟,只要研究通电时的电子学问题就可
以了,这样,可以利用已知的电子学知识来解决问题,根据各电极基本过
程对时间的不同响应,可以使复杂的等效电路得以简化或进行解析,从而
简化问题的分析和计算。
1. 暂态过程的分析方法
12
电极过程的等效电路以及等效电路之间的关系,可以根据各个电极基本过程的
电流、电位之间的关系来确定。
电荷转移过程的等效电路
电极界面上规则排布着异种电荷,形成界面双电层,这一双电层非常类似于
一个平板电容器,等效成一个双电层电容,用Cd表示。
Cd
S M
3
13
电极界面上还进行电荷转移过程,电荷转移的速度由法拉第电流来描述。如果电
荷转移过程比较慢,法拉第电流会引起电化学极化的过电位,这一电流、电位关
系非常类似于一个电阻上的电流、电压关系,因此,这时的电荷转移过程可以等
效成一个电阻,称为电荷转移电阻或电化学反应电阻,用Rct表示。
Rct
M+
M+
M+
e-
e-
e- (M++e-→M)
S M
电荷转移过程的等效电路
14
在暂态过程中,总的极化电流等于双电层电容的双电层充电电流ic和流过反应
电阻Rct的法拉第电流if之和,即
c fi i i
而且,反应电阻Rct两端的电压(即电化学极化过电位)正是通过改变双电层荷电
状态建立起来的,就等于双电层电容Cd两端的电压。综合考虑Cd和Rct之间的电
流、电压关系,可知Cd和Rct之间应该是并联关系。
Cd
Rct
ic
if
ηe
电荷转移过程的等效电路
15
当极化电流通过电极/溶液界面时,电化学反应开始,这样会导致界面上反应物
的消耗和产物的积累,出现了浓度差。通电初期,扩散层较薄,扩散速度较快
,因此没有浓差极化出现。随时间的推移,扩散层逐步向溶液内部发展,扩散
速率减慢,浓差极化开始建立并逐渐增大。当扩散达到对流区时,电极进入稳
态扩散状态,建立起稳定的浓差极化过电位。因此,浓差极化过电位的出现和
增大是逐步的、滞后于电流的,这个电压、电流的关系很像含有电容的电路两
端的电压、电流的关系。
扩散过程的等效电路
Rw Cw
Zw
为了给一个完整的概念,一般用一个半无限扩散阻抗Zw来表示扩散过程的等效电路:
16
扩散传质过程和电荷转移过程是连续进行的,两个过程的速度相等,因此,两
个过程的等效电路上流过的电流均为法拉第电流,同时,界面极化过电位是由
浓差极化过电位和电化学极化过电位组成,也就是说,扩散阻抗两端电压和反
应阻抗两端电压之和为总电压。很明显,它们应该是串联关系,总阻抗为法拉
第阻抗(Zf)。
总的极化电流等于流过双电层电容的充电电流和流过法拉第阻抗的电流之和,
而且,法拉第阻抗两端的电压是通过改变双电层荷电状态建立起来的,就等于
双电层两端的电压,由此可见,Cd和Zf之间应该是并联关系。
整个界面等效电路如下图:
Cd
Rct Zw
扩散过程在电极等效电路中的位置
17
流过电极的极化电流除了流经界面,还流过溶液和电极,极化电流在从参比电
极的Luggin毛细管管口到研究电极表面之间的溶液电阻Ru上产生的溶液欧姆压
降,因此,这段溶液电阻和界面等效电路串联,构成了总的电极等效电路。
暂态过程的等效电路
Cd
Rct
Zw
Ru
上述等效电路是具有四个电极基本过程(双电层充电、电荷转移、扩散传质和离子
导电过程)的等效电路,电路中的四个元件分别对应电极过程的四个基本过程。
18
根据施加电信号的不同:控制电流、控制电位和控制电量方法。在控制电流
和控制电量的暂态测量技术中,测量的响应信号为电位;而在控制电位的暂
态测量技术中,测量的响应信号为电流。
按照极化方式的不同:阶跃法、方波法、线性扫描法和电化学阻抗法。
在阶跃扰动时,电极电位或流过电极的外测电流被突然控制为一个预设的
恒定值,并保持该值不变,因此电化学反应系统可能逐渐趋近于新的稳态;
而在持续扰动时,由于电极电位或流过电极的外测电流不断变化,因此体
系可能一直无法达到稳态。
暂态法的分类
当电化学反应体系处于暂态,就可进行暂态测量,暂态来源于电极过程条件
的改变,这种改变既可以来自于过程本身的变化,也可以来自于外加扰动。
我们所讨论的暂态测量技术,指的是对电极过程施加一定的电信号扰动,并
测量体系的电信号响应的技术。
4
19
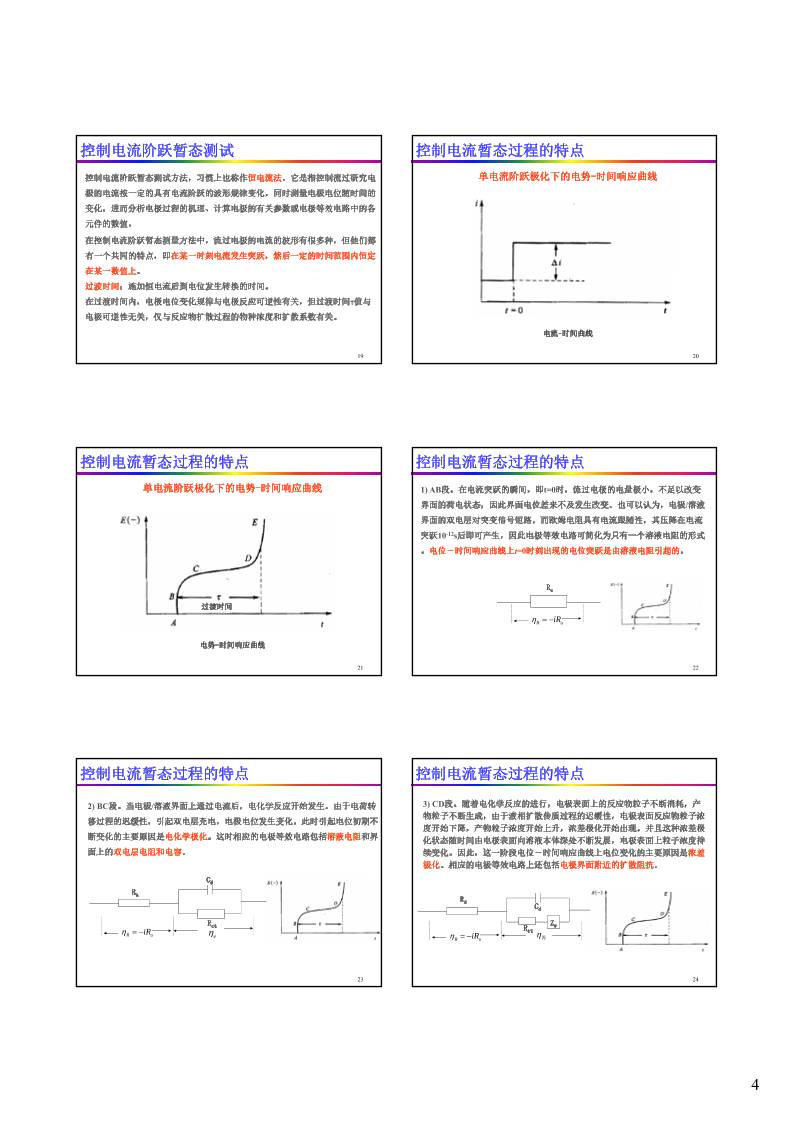
控制电流阶跃暂态测试方法,习惯上也称作恒电流法。它是指控制流过研究电
极的电流按一定的具有电流阶跃的波形规律变化,同时测量电极电位随时间的
变化,进而分析电极过程的机理、计算电极的有关参数或电极等效电路中的各
元件的数值。
控制电流阶跃暂态测试
在控制电流阶跃暂态测量方法中,流过电极的电流的波形有很多种,但他们都
有一个共同的特点,即在某一时刻电流发生突跃,然后一定的时间范围内恒定
在某一数值上。
过渡时间:施加恒电流后到电位发生转换的时间。
在过渡时间内,电极电位变化规律与电极反应可逆性有关,但过渡时间τ值与
电极可逆性无关,仅与反应物扩散过程的物种浓度和扩散系数有关。
20
控制电流暂态过程的特点
单电流阶跃极化下的电势-时间响应曲线
电流-时间曲线
21
控制电流暂态过程的特点
单电流阶跃极化下的电势-时间响应曲线
电势-时间响应曲线
过渡时间
22
1) AB段。在电流突跃的瞬间,即t=0时,流过电极的电量极小,不足以改变
界面的荷电状态,因此界面电位差来不及发生改变。也可以认为,电极/溶液
界面的双电层对突变信号短路。而欧姆电阻具有电流跟随性,其压降在电流
突跃10-12s后即可产生,因此电极等效电路可简化为只有一个溶液电阻的形式
。电位-时间响应曲线上t=0时刻出现的电位突跃是由溶液电阻引起的。
Ru
R uiR
控制电流暂态过程的特点
23
2) BC段。当电极/溶液界面上通过电流后,电化学反应开始发生。由于电荷转
移过程的迟缓性,引起双电层充电,电极电位发生变化。此时引起电位初期不
断变化的主要原因是电化学极化。这时相应的电极等效电路包括溶液电阻和界
面上的双电层电阻和电容。
R uiR
Ru
Cd
Rct
e
控制电流暂态过程的特点
24
3) CD段。随着电化学反应的进行,电极表面上的反应物粒子不断消耗,产
物粒子不断生成,由于液相扩散传质过程的迟缓性,电极表面反应物粒子浓
度开始下降,产物粒子浓度开始上升,浓差极化开始出现。并且这种浓差极
化状态随时间由电极表面向溶液本体深处不断发展,电极表面上粒子浓度持
续变化。因此,这一阶段电位-时间响应曲线上电位变化的主要原因是浓差
极化。相应的电极等效电路上还包括电极界面附近的扩散阻抗。
R uiR
Ru
Cd
Rct
Zw
界
控制电流暂态过程的特点
5
25
4) DE段。随着电极反应的进行,电极表面上反应物粒子的浓度不断下降,当
电极反应持续一段时间后,反应物的表面浓度下降为零,达到了完全浓差极化
。此时,电极表面上已无反应物粒子可供消耗,在恒电流的驱使下达到电极界
面上的电荷不能再被电荷转移过程所消耗,因此改变了电极界面上的电荷分布
状态,也就是对双电层进行快速充电,电极电位发生突变,直至达到另一个电
荷转移过程发生的电位为止。
控制电流暂态过程的特点
26
从上述分析可以看出,欧姆极化(电阻极化)、电化学极化和浓差极化这三种极
化对时间的响应各不相同。欧姆极化响应最快,电化学极化响应次之,浓差极
化响应最慢。换言之,电极极化建立的顺序是:欧姆极化、电化学极化和浓差
极化。
由于三种极化对时间的响应不同,因此我们可以通过控制极化时间的方法使等
效电路得以简化,突出某一电极基本过程,从而对其进行研究。
控制电流暂态过程的特点
27
控制电势暂态测试方法,习惯上也称作恒电势法。它是指控制电极电势按一定
的波形规律变化,同时测量电流随时间的变化,或者测量电量随时间的变化。
控制电势暂态测试方法
1) 电极界面电势差的变化会经历一个过程
a. 溶液欧姆电阻的存在。电势突变的瞬间,发生突变的是溶液的欧姆压降ηR
,而界面电势差来不及变化,瞬间电流达到-η/Ru,接着双电层充电,界面
电势差的绝对值逐渐增大,溶液欧姆压降的绝对值逐渐减小,在这段时间
内,界面电势差为体系总的过电势同溶液欧姆压降之差,并且逐渐逼近所
控制的总的过电势。
b.恒电势仪的输出能力有限。由于恒电势仪输出电流的限制,电势阶跃瞬间
双电层的充电电流不可能达到无穷大,因此双电层充电需要一定的时间。
当电极上施加一个电势突跃信号η(η为负值)进行极化的时候,虽然对电极体系
加上了一个电势差,但是电极溶液界面上的电势差(界面过电势)η界并不能立即
发生突跃。
控制电势阶跃暂态过程的特点
28
如果界面电势差可以在瞬间突跃到预定值,即
而
那么必然有iC→∞。
但由于恒电势仪的输出电流是一个有限值,不可能提供无穷大的双电层充电
电流,所以这是不可能的。也就是说,改变界面电势差所需要的双电层充电
过程需要一定的时间,不可能在瞬间完成,这样界面电势差的改变也不可能
在瞬间完成。因此,从极化开始到界面上稳定的电极电势的建立,必然经历
一个过渡阶段。
dE
dt
c d
dE
i C
dt
控制电势阶跃暂态过程的特点
29
控制电势阶跃极化时电极体系各部分超电势的变化趋势
在电势突跃的瞬间,发生突变的不是界面电势差(界面过电势),而是研究电
极与参比电极间的溶液欧姆压降,瞬间电流达到-η/Ru。接着,双电层被此电
流充电而发生电势变化,界面过电势开始建立,其绝对值不断增大。因为控
制的总过电势η是不变的,所以随着-η界的增大,-ηR不断减小。
控制电势阶跃暂态过程的特点
30
2) 电流的变化过程:总电流不断减小
Zw
i
ic
if
Ru
Cd
Rct
R 界
通过电极的总电流i随着-ηR的减小而不断减小。因为-η界不断增大,电化学反应
的电流if就不断增大,而总电流i是不断减小的,由i=iC+if可知,双电层充电电流ic
必然是不断减小的。电极界面上逐步建立的过电势η界包括电化学极化过电势,
随时间的延长还可能逐步建立起浓差极化过电势。当电极过程达到稳态时,双电
层充电过程结束,ic=0,i=if。在此过程中总的电流通常是不断减小的。
控制电势阶跃暂态过程的特点
6
31
控制电势阶跃极化时电极体系的响应电流曲线
32
循环伏安法
控制研究电极的电势以速率v从Ei开始向电势负方向扫描,到时间t=λ(相应电
势为Eλ)时电势改变扫描方向,以相同的速率回扫至起始电势,然后电势再次
换向,反复扫描,即采用的电势控制信号为连续三角波信号,记录i-E曲线,
称为循环伏安曲线。这一方法称为循环伏安法。
33
对于一个电化学反应 O ne R
O ne R
R O ne
正向扫描(即向电势负方向扫描)时发生阴极反应
反向扫描时,则发生正向扫描过程中生成的反应产物R的重新氧化的反应
循环伏安法
34
当从一个不发生电极反应的初始电势开始扫描时,暂态只有非法拉第电流流过
。随着电极电势逐渐负移到 (还原电位)附近时,O开始在电极上还原,并
有法拉第电流通过。由于电势越来越负,电极表面反应物O的浓度逐渐下降,
因此向电极表面的流量和电流就增加。当O的表面浓度下降到近于零,电流也
增加到最大值Ipc,然后电流逐渐下降。当电势达到r后,又改为反向扫描。
0
平
循环伏安法
35
随着电极电势逐渐变正,电极附近可氧化的R粒子的浓度较大,在电势接近
并通过 时,表面上的电化学平衡应当向着越来越有利于生成O的方向发
展。于是R开始被氧化,并且电流增大到峰值氧化电流Ipa,随后又由于R的
显著消耗而引起电流衰降。整个曲线称为“循环伏安曲线”。
0
平
循环伏安法
36
循环伏安曲线上有两组重要的测量参数:
a. 阴、阳极峰值电流IPc、IPa及其比值 |IPc/IPa|
b. 阴、阳极峰值电势差值|∆EP|=EPa-EPc
循环伏安法
7
37
对于产物稳定的可逆体系,循环伏安曲线两组参数具有下述重要特征:
a. |IPa|=|IPc|,即 且与扫速v、换向电势Eλ、扩散系数D等参数无关;
b.
可逆体系
1pa
pc
i
i
2.3 59
25o
p pa pc p pa pc
RT
E E E E E E mV
nF n
或 ( C)
38
准可逆体系循环伏安曲线两组参数的特征为:
a.
b. 准可逆体系的|∆Ep|比可逆体系的大,即
且随着扫速v的增大而增大。
准可逆体系
pa pci i
59
25o
p pa pcE E E mV
n
( C)
39
|∆Ep|值以及|∆Ep|随扫描速率v的变化特征是判断电极反应是否可逆和不可逆程
度的重要判据。
不可逆体系
2.3
p
RT
E
nF
如果 且不随v变化,说明反应可逆。
2.3
p
RT
E
nF
如果 且随v增大而增大,则为不可逆反应。
2.3
p
RT
E
nF
比 大得越多,反应的不可逆程度就越大。
不可逆体系进行单程线性电势扫描时,随着扫描速率v的增大,峰值电势向扫描
的方向移动,即阴极电势Epc向电势负方向移动,阳极峰电势Epa向电势正方向移
动,因此|∆Ep|随扫描速率增大而增大。
40
完全不可逆体系
当电极反应完全不可逆时,逆反应非常迟缓,正向扫描产物来不及发生反应就扩
散到溶液内部,因此在循环伏安图上观察不到反向扫描的电流峰。
41
初步研究电极体系可能发生的电化学反应
线性电势扫描伏安法常被用来研究一个未知电极体系可能发生的电
化学反应。
伏安曲线上出现阳极电流峰表示电极发生了氧化反应,阴极电流峰
表明发生了还原反应;
电流峰对应的电势范围可用于判定发生了什么电化学反应,与该反
应的平衡电势之间的差值表明了该反应发生的难易程度。
一对可逆反应对应的阴阳极电流峰的峰值电势差值表明了该反应的
可逆程度。
峰值电流则表示在给定的条件下该反应可能的进行速度。
42
初步研究电极体系可能发生的电化学反应
如果不存在干扰的话,对于给定的电极体系,在控制电势扫描的情况下
,相同的电极反应应该发生在相同的电势下,并以同样的速度进行。在
多次的循环伏安扫描过程中,如果电流峰的峰值电势或峰值电流随扫描
次数而发生变化往往预示着电极表面状态在不断变化。
如果把电流-电势曲线转换成电流-时间曲线,则电流峰下覆盖的面积
就代表该电化学反应所消耗的电量,由此电量可以得到电极活性物质的
利用率、电极表面吸附覆盖度、电极真实电化学表面积等一系列信息。
循环伏安法是定性或半定量的研究电极体系可能发生的反应及
其进行速度的首选方法。
8
43
电化学阻抗谱的特点:
1.一种以小振幅的正弦波电势(或电流)为扰动信号的电化学测量方法:
(1) 避免对体系产生大的影响
(2) 使扰动与体系的响应之间近似呈线性关系
2.一种频率域的测量方法:
以测量得到的频率范围很宽的阻抗谱来研究电极系统,速度快的子过程出现在
高频区,速度慢的子过程出现在低频区,可判断出含几个子过程,讨论动力学
特征。
电化学阻抗谱的解析:
最常用的分析方法是曲线拟合法。对电化学阻抗谱进行曲线拟合时,必须首先
建立电极过程合理的物理模型和数学模型,该物理模型和数学模型可揭示电极
反应的历程和动力学机理,然后进一步确定数学模型中待定参数的参数值,从
而得到相关的动力学参数或物理参数。用于曲线拟合的数学模型分为两类:一
类是等效电路模型,等效电路模型中的待定参数就是电路中的元件参数,另一
类是数学关系式模型。等效电路模型更常被采用。
五、电化学阻抗谱法
44
电化学阻抗谱的类型:
复平面图:以阻抗的实部为横轴,虚部为纵轴,也叫Nyquist plot。
波特图:Bode plot,由两条曲线组成,一条是描述阻抗的模随频率的变化关系,
即log|Z|-logf,称为Bode模图;另一条是描述阻抗的相位角随频率的变化关系,
即φ-logf,称为Bode相图。两条曲线要同时给出,才能完整描述阻抗的特征。
0.1 0.2 0.3 0.4 0.5 0.6 0.7 0.8 0.9
0.0
0.1
0.2
0.3
0.4
0.5
0.6
-Z
Im
(
)
Z
Re
()
1E-3 0.01 0.1 1 10 100 1000
0.1
0.2
0.3
0.4
0.5
0.6
0.7
0.8
0.9
IZ
I (
)
Frequency (Hz)
1E-3 0.01 0.1 1 10 100 1000
-25
-20
-15
-10
-5
0
T
h
e
ta
Frequency (Hz)
50%DOD, 10KHz-5mHz
五、电化学阻抗谱法
45
当对电池中的某一电极进行EIS测试时,可以得到电极内各组成部分对电极性能
的影响信息。下图所示阻抗谱是嵌入型电极上测得的典型阻抗谱,图中的标注是
引起相应频率范围阻抗响应的电极弛豫过程。
电化学阻抗的应用
若对锂离子电池的正、负极进行EIS测试,均可得到类似的电化学阻抗谱。通常
采用的测试频率范围为10-2~105 Hz,所得阻抗包括两个容抗弧和一条倾斜角度接
近45o的直线。 46
电化学阻抗的应用
下图是尖晶石锂锰氧化物正极在首次脱锂(充电)过程中不同电势下的电化学阻抗
谱。
图(a) 给出了尖晶石锂锰氧化物正极在开路电势3.5 V(vs. Li+ /Li )下的阻抗谱,谱
图高频区域存在一个小的容抗弧,中低频区域存在一段不完整的大容抗弧。高频
容抗弧对应着锂锰氧化物表面上覆盖的Li2CO3原始膜的弛豫过程,而中低频容
抗弧则对应着双电层电容通过传荷电阻的充放电过程,由于在此电势下脱锂过程
尚未发生,传荷电阻很大,因而此时中低频容抗弧很大。
47
图(b)给出了尖晶石锂锰氧化物正极在4.1 V ( vs. Li+ /Li )极化电势下的阻抗谱,谱
图高频区域存在一个较小的容抗弧,中低频区域存在一个较大的容抗弧,低频区
域则是一条倾斜角度接近45o的直线。当电极电势大于3.8V ( vs. Li+ /Li ),正极开
始充电后,阻抗谱均为由两个容抗弧和一条倾斜角度接近45o的直线构成。
电化学阻抗的应用
48
电化学阻抗的应用
大量关于嵌入型电极的研究表明,在电极表面存在着一层有机电解液组分分解形
成的,能离子导电而不能电子导电的绝缘层,称为固体电解质相界面(solid
electrolyte interphase,SEI)膜。SEI膜最早是在锂离子电池碳负极上发现的,近
几年的研究表明,SEI膜也存在于所有LixMOy(M=Ni、Co、Mn等)正极表面
上。因此,在锂离子电池充放电时,锂离子迁移通过SEI膜,到达或离开电极活
性材料表面的过程,是整个电极过程的一个组成部分。
(b)图中阻抗谱的高频容抗弧对应着锂离子在SEI膜中的迁移过程,而中频容抗弧
则对应着锂离子在SEI膜和电极活性材料界面处发生的电荷传递过程,低频直线
对应着锂离子在固相中的扩散过程。电极的等效电路图如下:
9
49
图中,R代表电极体系的欧姆电阻,包括隔膜中的溶液欧姆电阻和电极本身的
欧姆电阻;常相位元件QSEI和RSEI分别代表SEI膜的电容和电阻;常相位元件Qd
代表双电层电容;Rct代表电荷传递电阻;常相位元件Qw代表固相扩散阻抗。
电化学阻抗的应用
前面所述等效电路的对4.1V ( vs. Li+/Li)极化电势下的阻抗谱进行曲线拟合,可以
获得良好的拟合效果,2值为7.18×10-4,拟合所得元件参数列于下表:
50
六、 电势阶跃法
电势阶跃法是给研究电极施加一个电势突跃信号η,电极进行强极化,记录电
流随时间的变化关系。
对一还原反应,电势阶跃法是将电势从不发生反应的φ1阶跃至较负的电势φ2时
,发生非常迅速的还原反应,以至于扩散到电极表面的物质将立即反应,使电
极表面的物种浓度始终为0,这时测得的电流是极限扩散条件下的电流,电流和
时间的关系符合Cottrell方程:
1/2 1/2 0 1/2( )di t nFA D C t
n为在氧化还原反应中的得失电子数,F为Faraday常数,C0为氧化物物种的起
始浓度。
由此可见,从电流与时间的关系可以得到反应的扩散系数D值。
51
0 200 400 600 800 1000 1200 1400 1600 1800 2000
-3.5
-3.0
-2.5
-2.0
-1.5
-1.0
-0.5
0.0
lo
g
(i
)
(i
(
A
/g
))
Time (s)
上图为满充状态的贮氢合金电极在+600 mV电势阶跃后的阳极电流与时间之间的响应曲
线。从图中可以看出,logi – t曲线可以明显的分为两个阶段,一是放电初始阶段,在这
一阶段满充状态的合金电极由于加载了一个较大的阳极电势阶跃(形成一个很大的过电
势),合金电极表面的电荷转移速度非常快,合金电极表面的氢浓度接近于零,阳极电
流迅速下降,这一阶段由于反应较快,持续时间较短;
52
0 200 400 600 800 1000 1200 1400 1600 1800 2000
-3.5
-3.0
-2.5
-2.0
-1.5
-1.0
-0.5
0.0
lo
g
(i
)
(i
(
A
/g
))
Time (s)
二是经过足够长的时间后,氢在合金体相内的扩散成为恒电势放电的速率控制步骤,阳
极电流缓慢下降, logi与t之间呈现出良好的线性关系。根据球形扩散理论,当放电时间
足够长时,扩散电流与时间的关系遵循如下公式:
2
02 2
6
log log( ( ))
2.303s
FD D
i C C t
da a
通过实验作出logi与t的关系曲线,再进行线性拟合即可得到D/a2值,根据测得的合金颗
粒半径,即可求出氢在合金体相内的扩散系数D。 D=1.47×10-10 cm2/s
高速下载:
点击后进入安全下载页,再进行实际下载。下载链接有效期 24 小时,过期会自动刷新。




 WMS仓库系统
WMS仓库系统